第八章 休 克
概述
※休克(shock):是各种强烈致病因子作用于机体引起的急性循环衰竭,其特点是微循环障碍,重要脏器灌流的不足和细胞代谢障碍,由此引起的全身性危重的病理过程。【重点】
正常微循环的结构和生理特点
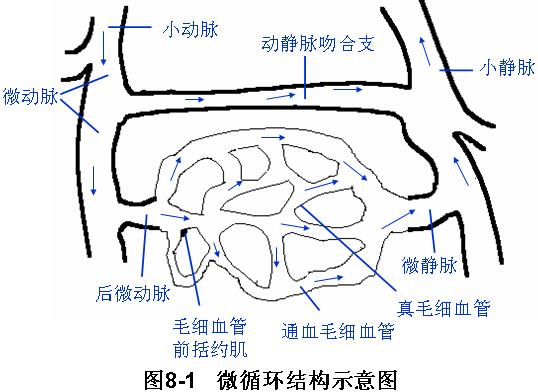
(一) 微循环的组成和功能
(二)微循环通路
1、迂回通路:血液流经微动脉、后微动脉、毛细血管括约肌到微静脉。
2、直接通路:血液流经微动脉、后微动脉、通血毛细血管到微静脉。
3、动-静脉短路:血液由微动脉经动-静脉吻合支到微静脉。

组织灌流量:主要是指单位时间内流经真毛细血管的血流量。 组织灌流量,组织有效灌流量,微循环灌流量是同义语。
(三)影响组织灌流量的因素
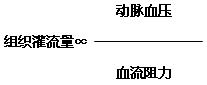
组织灌流量和动脉血压成正比,与微循环的血流阻力成反比。
1、毛细血管前阻力(ra):将微动脉、后微动脉、毛细血管前括约肌的舒缩对血流构成的阻力称毛细血管前阻力。毛细血管前阻力主要影响微循环血液的灌入。
2、毛细血管后阻力(rv):将微静脉、后微静脉的舒缩对血流构成的阻力称毛细管后阻力。毛细管后阻力主要影响微循环血液的流出。

四、微循环的调节

第一节 病因和分类
(一)失血性和失液性休克
(二)烧伤性休克
(三)创伤性休克
(四)感染性休克
(五)过敏性休克
(六)神经源性休克
(七)心源性休克
二、按发生休克的起始环节分类
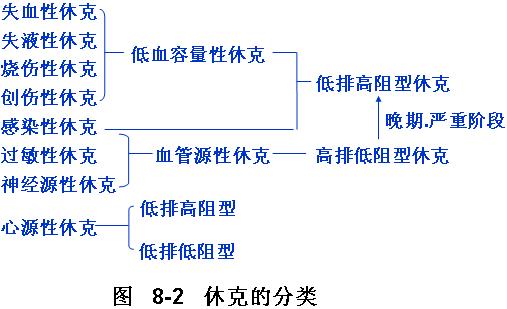
★《按休克的起始环节可将休克分成三种类型:低血容量性休克、血管源性休克、心源性休克其起始环节、发生休克的机制是:》【重点】
(一)低血容量性休克:由于血量减少引起的休克。起始环节是血容量减少。由于血容量减少,导致静脉回流不足,心输出量下降,血压下降,由于减压反射受抑制,交感神经兴奋,外周血管收缩,组织灌流量进一步减少。
(二)血管源性休克:由于血管扩张,血管床容量增加,造成有效循环血量相对不足,使组织灌流量减少引起的休克。起始环节是血管床容量增加。休克时由于组织长期缺血、缺氧、酸中毒、和组胺及一氧化氮等活性物质的释放,造成血管张力的低下,通透性增加,加之白细胞、血小板在微静脉端粘附,造成微循环血液淤滞,毛细血管开放数增加,导致有效循环血量锐减。
(三)心源性休克:由于急性心泵功能障碍或严重的心律紊乱而导致的休克。起始环节是心输出量急剧减少。由于心输出量迅速降低,血压可显著降低,多数病人外周阻力增高(低排高阻型)这是因为血压降低,使主动脉弓和颈动脉窦压力感受器的冲动减少,反射性引起交感神经传出冲动增多,引起外周小动脉收缩,使血压能有一定程度的代偿。少数病例外周阻力降低(低排低阻),这是由于这类病人心肌梗塞面积大,心输出量显著降低,血液淤滞在心室,使心室壁牵张感受器牵拉,反射性地抑制交感中枢,使交感神经传出的冲动减少,外周阻力降低,引起血压进一步下降。
三、按休克的血液动力学特点分类
(一)感染性休克的分类
1、高动力型休克(高排低阻性休克)
血液动力学特点:心输出量增加,外周阻力降低。
2 、低动力型休克(低排高阻型休克)
血液动力学特点:心输出量减少,外周阻力增高。
(二)心源性休克的分类
1、低排高阻型
2、低排低阻型
第二节 休克的分期与发病机制
一、缺血性缺氧期(休克早期)
★《休克的缺血性缺氧期微循环的改变及其机制》【重点】
本期微循环变化的特点是缺血。
1、 微循环缺血
休克早期的全身小血管,包括小动脉,微动脉,后微动脉,毛细血管前括约肌和微静脉,小静脉都持续痉挛,使毛细血管前阻力和后阻力都增加,由于微静脉、小静脉对儿茶酚胺更敏感,其中主要以毛细血管前阻力增加显著,同时大量真毛细血管网关闭,开放的毛细血管减少,毛细血管血流限于直接通路,动静脉短路开放,组织灌流量减少,微循环出现少灌少流,灌少于流的情况。

2.微循环缺血的机制
(1)、交感-肾上腺髓质系统的强烈兴奋:不同类型的休克可通过不同的机制引起交感-肾上腺髓质系统的兴奋,例如:低血容量性休克和心源性休克由于血压低,减压反射被抑制,引起心血管运动中枢及交感-肾上腺髓质系统兴奋,儿茶酚胺大量释放,使小血管收缩;烧伤性休克时疼痛刺激可引起交感-肾上腺髓质系统兴奋,血管收缩比单纯失血为甚;在败血症休克时,内毒素有拟交感神经系统的作用。交感-肾上腺髓质系统的强烈兴奋使儿茶酚胺大量释放,即刺激α受体,造成皮肤,内脏血管明显痉挛,又刺激β-受体,引起大量动静脉短路开放,构成了微循环非营养性血流通道,使器官微循环血液灌流锐减。
(2)、休克时体内产生一些具有促使血管收缩作用的体液因子。如血管紧张素Ⅱ、加压素、血栓素、内皮素、心肌抑制因子,及白三烯类物质等都具有促进血管收缩的作用。
2、 代偿意义
★《休克的缺血性缺氧期微循环变化的代偿意义》【重点】
(1)、自身输血:肌性微静脉和小静脉收缩,肝储血库收缩,减少血管床容量,可以迅速而短暂地增加回心血量这种代偿起到“自身输血”的作用,是休克时增加回心血量的“第一道防线”。
(2)、组织液返流入血:由于毛细血管前阻力大于毛细血管后阻力,毛细血管中流体静压下降,使组织液进入血管中,起到“自我输液”的作用。这是休克时增加回心血量的“第二道防线”。
(3)、钠水重吸收增加:RAS的激活,醛固酮分泌增多及ADH分泌增多,肾对钠水重吸收增加,使有效循环血量增加。
(4)、血液重新分布:
由于不同器官的血管对儿茶酚胺反应不一,皮肤,内脏、骨骼肌,肾的血管α受体密度高,对儿茶酚胺敏感性较高,血管收缩强烈,而脑血管α受体密度低,则无明显改变。CA作用冠状动脉β受体使其舒张。这种血流重分布,保证了心、脑主要生命器官的血液供应。
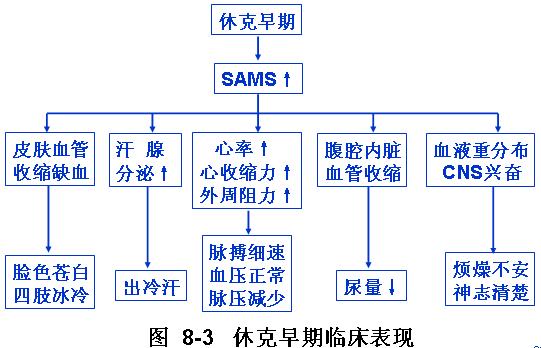
(5)、心输出量增加及外周阻力升高:SAMS兴奋,使心率加快,心收缩力增强,心输出量增加及外周阻力增加,减轻了血压的下降的程度,有利于动脉血压的维持。所以早期休克的病人血压可正常,神志清楚。
4.临床表现
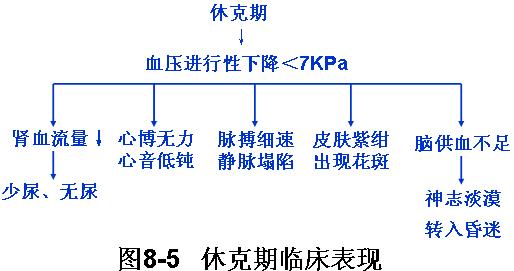
二. 淤血性缺氧期(休克期)
★《休克的淤血性缺氧期微循环的改变及其机制》【重点】
本期微循环变化的特点是淤血。
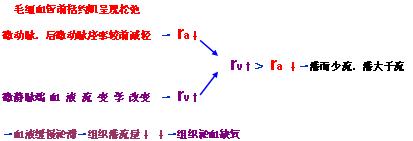
1. 微循环淤血
(1)毛细血管前阻力降低:休克的淤血性缺氧期,内脏微循环中的血管运动现象首先消失,终末血管床对儿茶酚胺的反应性降低,此时,血液不再局限于通过直捷通路,而是经过毛细血管前括约肌大量涌入真毛细血管网,此时微动脉和后微动脉痉挛较前减轻,使毛细血管前阻力降低。
(2)毛细血管后阻力升高:由于微静脉端的血流缓慢,红细胞发生聚集,白细胞滚动、粘附、贴壁嵌塞,血小板聚集,血粘度增加,微血流流态的改变,使毛细血管后阻力增大。
由于毛细血管的后阻力大于前阻力,组织灌而少流,灌大于流,真毛细血管开放数目增多,血流更慢,甚至淤滞,组织处于严重的淤血缺氧状态。
1. 微循环淤血的机制
(1)、酸中毒:长时间的缺血和缺氧引起组织氧分压下降,CO2和乳酸堆积,发生酸中毒。酸中毒导致血管平滑肌对儿茶酚胺的反应性降低。
(2)、局部的代谢产物增多:长时间的缺血缺氧引起局部血管扩张,代谢产物增多,如释放组胺增多,ATP的分解产物腺苷和细胞分解时释放的K+增多,组织间液渗透压增高,这些都可以造成血管扩张。(3)、内毒素作用:除病原微生物感染引起的败血症外,休克后期常有肠源性细菌和脂多糖(LPS)入血。LPS和其它毒素可以通过多种途径,引起血管扩张,持续性低血压。
(4)、血液流变学(hemorheology)的改变:近年研究表明,血液流变学的改变在休克期微循环淤血的发生发展中起着非常重要的作用。休克期白细胞滚动,贴壁,粘附于内皮细胞上,加大了毛细血管的后阻力。血液浓缩,血细胞压积上升,血液粘滞度增大,红细胞聚集,血小板粘附聚集,都造成微循环血流变慢,血液泥化,淤滞,甚至血流停止。
●《休克的淤血性缺氧期血液流变学改变及其变化的机制》【难点】
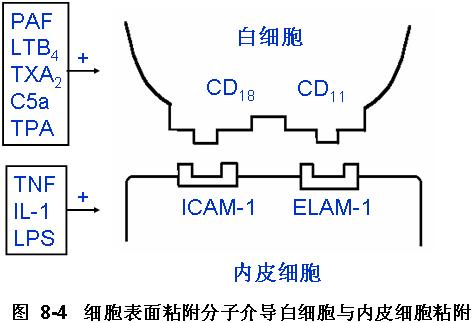
休克期白细胞滚动,贴壁,粘附于内皮细胞上,加大了毛细血管的后阻力。这种粘附是受细胞表面粘附分子所介导的。参加血细胞粘附的细胞粘附分子(Leu-CAMs)即CD11/CD18,存在于白细胞膜上,受PAF ,LTB4,C3a,C5a,TXA2及佛波醇脂激活后产生,细胞间粘附分子-1(intercellular adhesion molecule-1,ICAM-1)和内皮细胞-白细胞粘附分子(endothelial leukocyte adhesion molecule,ELAM)存在内皮细胞膜上,在TNF,IL-1及LPS,氧自由基刺激下产生,起着CD11/CD18粘附受体的作用,介导白细胞粘附,并激活白细胞引起微循环障碍及组织损伤。此外还有血液浓缩,血细胞压积上升,血液粘滞度增大,红细胞聚集,血小板粘附聚集,都造成微循环血流变慢,血液泥化,淤滞,甚至血流停止。
3、休克失代偿 休克期属失代偿期:
4、临床表现
二、 微循环衰竭期(休克晚期)
本期微循环变化的特点是广泛形成微血栓。
此期可发生DIC或重要器管功能衰竭,甚至发生多系统器官功能衰竭,成为休克难治期。
1、DIC形成
★(1)休克发生DIC的机制【重点】
①由于血液进一步浓缩,血细胞压积和纤维蛋白原浓度增加,血细胞聚集,血液粘度增高,加上血流速度显著变慢,酸中毒越来越严重,血液处于高凝状态。
②特别是败血性休克,感染的病原微生物与毒素均可损伤血管内皮,激活内源性与外源性凝血系统。
③严重的创伤性休克,组织因子入血,可启动外源性凝血系统。
④异型输血引起的溶血释放的红细胞素,也容易诱发DIC。
(2)微循环的变化
此时微循环内微血管扩张,有大量微血栓阻塞,微循环血流停止,不灌不流,组织得不到营养供应,微血管平滑肌麻痹,对任何血管活性药物失去反应。所以称微循环衰竭期。
●(3)DIC的严重影响【重点】
DIC一旦发生,将使微循环障碍更加严重,休克病情进一步恶化,这是因为:
(1)DIC时微血管阻塞了微循环通路,使回心血量锐减。
(2)凝血与纤溶过程中的产物,纤维蛋白肽和纤维蛋白降解产物,和某些补体成分,增加了血管的通透性,加重了微血管舒缩功能紊乱。
(3)DIC时出血。导致血量进一步减少,加重了循环障碍。
(4)器官栓塞、梗死,加重了器官急性功能衰竭。
2、 重要器官功能衰竭,甚至发生多系统器官功能衰竭。

第三节 休克的细胞代谢改变及器官功能障碍
一、细胞代谢障碍
(一)供氧不足,糖酵解加强
(二)能量不足,钠泵失灵
(三)局部酸中毒
二、细胞的损伤与凋亡
(一)细胞的损伤
1、细胞膜的变化
2、线粒体的变化
3、溶酶体的变化
(二)细胞凋亡
三、重要器官功能衰竭
各种类型休克常伴发急性肾功能衰竭称为休克肾。临床表现为少尿或无尿,氮质血症,高钾血症及代谢性酸中毒。
(二) 急性呼吸功能衰竭
休克肺(shock lung):因休克死亡的病人,尸检可见肺充血,水肿,出血,肺不张,肺泡内透明膜形成及肺毛细血管内微血栓形成等病理变化称为休克肺。【重点】
休克肺属于成人呼吸窘迫综合症(ARDS)之一。发生机制与休克动因通过补体―白细胞―氧自由基损伤呼吸膜有关。
休克肺的病理变化可引起肺的通气功能障碍,弥散障碍,肺泡通气/血流比例失调,进行性低氧血症和呼吸困难,从而导致急性呼吸衰竭甚至死亡。休克肺约占休克死亡人数的1/3。
(三) 心功能障碍
休克持续时间越久,心力衰竭越严重,可严重影响休克的予后。心力衰竭是休克患者三大死因之一。
★休克时心功能障碍的发生机制:【重点】
(1)冠状动脉血流量减少和心肌耗氧量增加;由于休克时,血压降低及心率加快所引起的心室舒张期缩短,可使冠状动脉灌流量减少和心肌供血不足;同时,交感-肾上腺系统兴奋,引起心率加快和心肌收缩加强,导致心肌耗氧量增加,因而加重了心肌缺氧。
(2)酸中毒和高钾血症使心肌收缩性减弱;
(3)心肌抑制因子(MDF)使心肌收缩性减弱;
(4)心肌内DIC形成,使心肌受损;
(5)细菌毒素(特别是革兰氏阴性细菌的内毒素),通过其内源性介质,引起心功能抑制。
(四)脑功能障碍
(五)消化道和肝功能障碍
●《几乎所有类型的休克晚期都可能引起内毒素血症》【重点】
休克时胃肠因缺血、淤血和DIC形成,发生功能紊乱。肠壁水肿,消化腺分泌抑制,胃肠运动减弱,粘膜糜烂,形成应激性溃疡,肠道细菌大量繁殖,肠粘膜屏障功能严重削弱,大量内毒素甚至细菌可以入血,从而使休克加重。
休克时肝缺血、淤血常伴有肝功能障碍,使由肠道入血的细菌内毒素不能充分解毒,引起内毒素血症。同时乳酸不能转化为葡萄糖或糖原,加重了酸中毒,这些改变都促使休克恶化。
